

Published on: October 4, 2023
Computational methods have been paving the way for the advent of PPIs prediction. The emergence and technological development of machine learning raise the possibility of more precise prediction of PPIs.
But, for the beginners of machine learning or scientists in other fields, it takes hard time to clearly understand the state-of-the-art computational methods. It often prevents positive attitudes toward predictive methodologies. In science, deep understanding of the assumptions and the theory is necessary to make a practical use to create a new technology. We need to catch up with the rapid progress of deep learning.
This short review in Proteomics provides us fairly comprehensive and easy-to-understand for the beginner.1) This review describes the methodological background of PPIs prediction from like a textbook. For example, the architecture of convolutional neural network (CNN), recurrent neural network (RNN), long short-term memory (LSTM), attention mechanism that led to the advent of transformer neural architecture as well as autoencoder are explained in the context of PPIs prediction pipeline development, The author takes several case examples of each architecture and it helps better understanding of basic methodologies of deep learning.
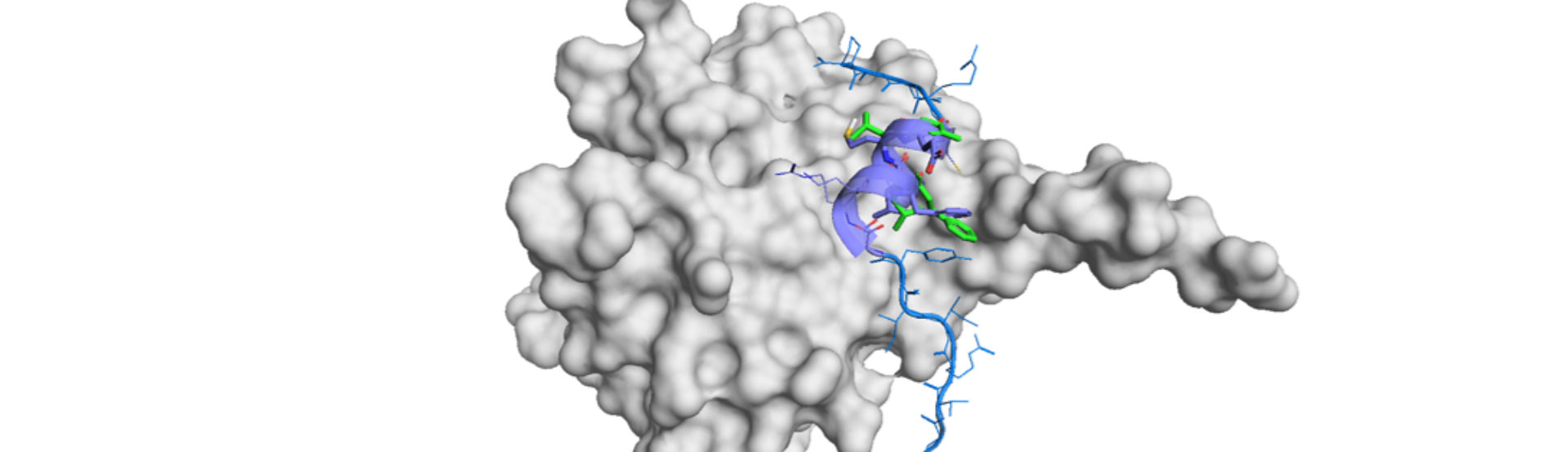
PPIs prediction is possible from one or more of the datasets of text-based primary structures, 3D PDB structures, metric extractions (amino acid composition, conjoint triad, auto covariance, etc.), biochemical co-fractionation to mass spectrometry (CF/MS) and so on. Let us introduce a case from the review article.
It would be desirable to predict a PPI precisely from the raw protein sequences. ctP2ISP is a CNN-based deep learning method that takes sample-oriented sampling strategy by data augmentation.
In ctP2ISP, PPI is predicted by the physicochemical properties of a pair of proteins. This method first picks up 500 residues and put into SPIDER3, a predictive method of protein secondary structure and its physicochemical properties like solvent accessibility area and backbone angles.3)
Data augmentation is achieved by definition of a local block against the global block. The global block refers to the 500-residue protein sequence and the local block uses 30 amino acid-fragment from the entire sequence. It allows more data generation for training and testing without accessing additional datasets.
Judging from the algorism of ctP2ISP, this pipeline is focusing on local PPI prediction rather than global one. Sacrificed sequence data could be covered by multiple trials against a series of 500 residues. But it is more practical to use this pipeline for the sake of the target-oriented and site-specific PPI prediction or PPI region identification.
In addition, ctP2ISP would have higher accuracy for secondary structure-based PPI because it is based on the physicochemical properties especially on the secondary structure-based ones. α-helices, β-sheets and β-turns are structurally well-fixed and these structure-based PPIs would show a good compatibility with this pipeline.
Deep understanding of PPIs predictions methods allows us to make a reasonable and reliable application of novel pipelines. This review would be a great introduction for the beginners of machine learning to construct the very basis.
Understanding is indispensable to use a methodology or to merge technologies. We are seeking for a partner in any field for innovation. Please contact us if you are interested and we would be glad to introduce you our technology more deeply.
1) https://doi.org/10.1002/pmic.202200292
2) https://doi.org/10.1109/tcbb.2022.3154413
3) https://doi.org/10.1093/bioinformatics/btx218