

Published on: July 10, 2024
Integration of technologies is a fascinating approach for innovation. A synergy of technologies in high compatibility would increase the success rate in drug design, discovery and development, and reduction of time and cost as well. We are eager to integrate novel technologies in our drug discovery process as the endeavor for the accuracy and acceleration.
An ensemble of different technology was mentioned in this blog before. Here is an report of different type of integration,1) which combines different in silico and machine learning (ML) technologies to furnish all-in-one basis of computational approach for hit discovery.
Basically, the workflow is 1) protein-protein interaction (PPI) interface analysis, 2) Virtual screening to identify the hit compound and 3) evaluation of molecular properties. The author utilizes the technologies below.
-AlphaSpace to target PPI interfaces2)
-Fpocket3) and MOE to predict the binding pocket
-Delta machine learning (-ML) scoring functions in virtual screening4)
-sPhysNet5) (simplified PhysNet6)) to predict molecular properties
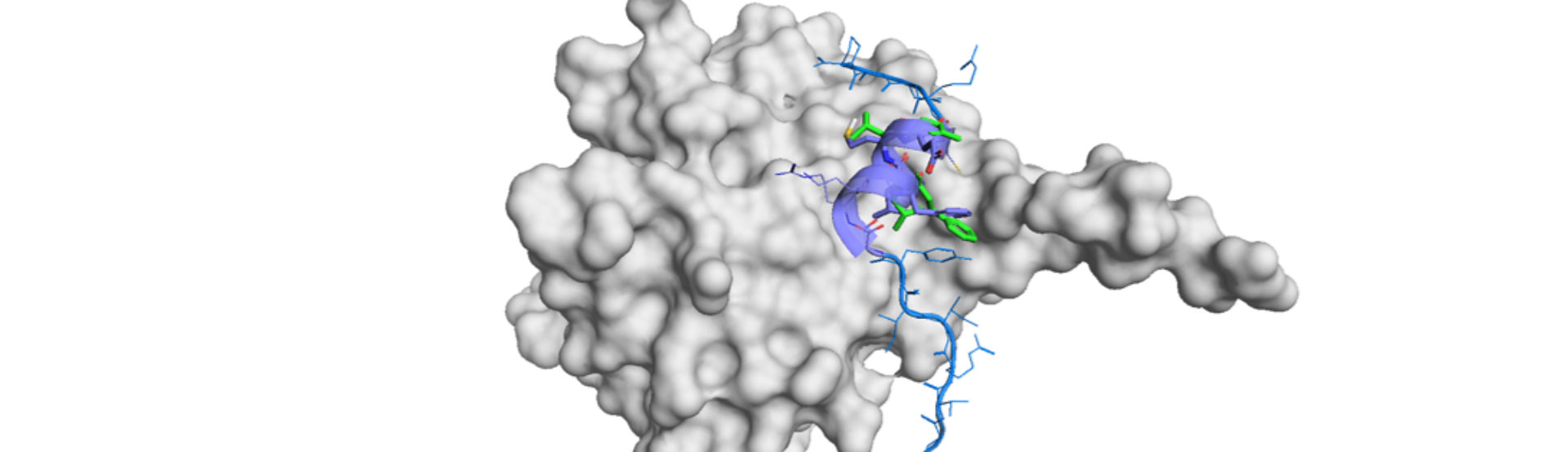
AlphaSpace is a tool for fragment-centric topographical mapping of PPI interfaces. It maps out concave interaction space at the protein surface using the alpha sphere and the Voronoi diagram.7) Fpocket and MOE’s SiteFinder to build up the central construct.
-ML scoring function utilizes ML to correct the gap between experimental and computational binding affinity for more accuracy. The author compares a series of ML models and find out unbiased RTMScore.8) RTMScore is advantageous because it adopts the probability density distribution of distances between ligand atoms and target amino acid residues.
sPhysNet is a biased protocol for deep neural network for the prediction of properties, including energies, forces and dipole moments. It basically employs the original concept of Phys Net and use MMFF for geometry optimization and DFT for energy optimization. But the dataset is uniformly biased: the available heavy atoms are limited to 20 and the input is fragment-based data.
The author specifically utilized the deep learning model called sPhysNet-MT-ens5.9) This model utilizes both calculated and experimental dataset and transfer learning in order to predict multiple parameters with higher accuracy. After the MMFF geometry is optimized based on the calculated dataset, sPhysNet-MT operates the prediction of electronic energies in gas, water and octanol levels. Then the transfer learning is performed to predict other parameters, like LogP and hydration free energy, from the experimental dataset.
The reliability of the integrated process was demonstrated for EGFR (PDB: 1M17). It is not the forward prediction to find our a more potent molecule, but still, the prediction output the similar experimental data for the EGFR kinase inhibitor, Erlotinib.
The authors’ integrated approach is one of the suitable ways for the interconnected process in drug discovery because the model could be expanded to work on other tasks of prediction as well. Tremendous amounts of tasks of drug discovery process hampers rapid candidate identification and validation. So long as fragment technologies are rapidly budding, integration opens up more possibility in the real application.
We are always welcome to push technology integration forward. PepMetics® molecules and our platform are useful for ML-based approaches, but also technologies related to PPIs. Please contact us if you are interested in integration of our technologies.
1) https://doi.org/10.1021/acs.jctc.3c00814
2) https://doi.org/10.1021/acs.jcim.5b00103
3) https://doi.org/10.1186/1471-2105-10-168
4) https://doi.org/10.1021/acs.jcim.2c00485
5) https://doi.org/10.1021%2Facs.jcim.1c00007
6) https://doi.org/10.1021/acs.jctc.9b00181
7) https://doi.org/10.1016/j.sbi.2004.03.010
8) https://doi.org/10.1021/acs.jmedchem.2c00991
9) https://doi.org/10.1021/acs.jctc.2c01024