

Published on: November 13, 2024
Prediction and selection of a ligand binding mode with high accuracy is still a difficult challenge in molecular docking even in this age of high computational capacity. Numerous challenges have been performed for over a century due to the overwhelming demand for protein-ligand complex in drug discovery. Induced fit is one of the intriguing but terrible phenomena in ligand-protein binding. Accurate prediction of induced fit would lead to increase in the value of molecular docking.
Induced fit is a conformational change of the protein by binding of the ligand. The concept of induced fit dates back to 1985.1) Before the discovery of induced fit phenomena, the protein was defined as a rigid entity. Generation of the ligand molecule conformation to score the binding modes was the main task of molecular docking.2)
This approach is computationally beneficial with the speediness of output. The hypothesis of rigidity reasonably fits and molecular docking results in reliable visualization of protein-ligand complex. However, it loses accuracy of prediction related to protein’s flexible conformational change.
Induced fit docking (IFD) has long been a problem to be overcome. QM/MM-based and HADDOCK-based approaches are relatively earlier ones that investigated the rationality and established unified approaches for general application.3),4) Schrödinger’s IFD-MD is one of the most successful cases that predicted 85% out of 258 protein- ligand pairs.5)
Here is a recent approach that built up the workflow of IFD by integration of CHARMM-GUI.6),7) CHARMM-GUI is a web-based platform for the inputs for molecular simulations. Its output is applicable for not only CHARMM but also AMBER, OpenMM, NAMD, GROMACS, Tinker, LAMMPS, GENESIS, LAMPS and Desmond.
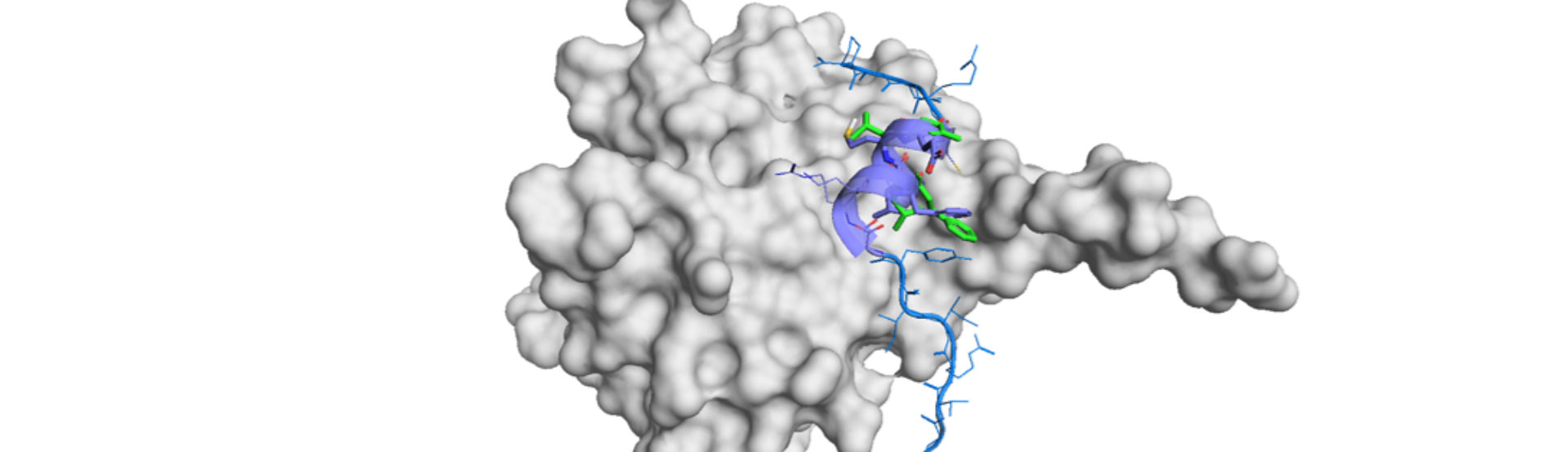
CHARMM-GUI-based IFD workflow (CGUI-IFD) comprises of two CHARMM-GUI modules: LBS-FR (Ligand Binding Site-Finder&Refiner) and HTS (High-Throughput Simulator). LBS-FR is a template-based generator of an ensemble of binding pocket conformations. The resultant rigid protein structures are used for molecular docking by HTS.
CHARMM-GUI HTS generates MD systems and their necessary input and evaluates binding pose and stabilities by the ligand RMSD and MMGBSA (molecular mechanics with the generalized Born surface area continuum solvation). This approach successfully predicted the binding modes in 80% using the same set with the demonstration by Schrödinger’s IFD-MD.
The authors evaluated the 20% of the failed cases. Increase in the number of templates for protein binding sites and ligand binding poses could be a rescue them and 8 test trials out of 10 samples had success in the reproduction of the protein-ligand binding mode. In the remaining failed cases, a large number of rotatable bonds of the ligand and complex hydrogen network in the protein pocket, respectively. The former problem happens with cross-docking approaches: requisite ligand conformation is not generated or highly scored by the algorism. The latter problem arose from the failure in generation of accurate pocket conformation without the ligand.
CGUI-IFD takes advantage of LBS-FR to generate a possible set of protein pocket conformation. It potentially includes the requisite conformation of the ligand-bound protein. But cross-docking is often suffered from failed generation of the protein pocket conformation and so for the ligand.
Approaches for IFD do not always imitate the process of induced fit. Induced fit is initiated by the contact or approach of a ligand to the protein. The ligand and the protein mutually interact and change the conformation to the optimal binding mode. This process is not simulated by cross-docking. It is understandable that prediction fails when the binding induces significant conformation change, where. the binding energy compensates the high energy conformation in the unbound environment.
The accuracy of IFD has been increased and now 80% or more cases would be predicted calculated. IFD-MD and CGUI-IFD are reliable when the binding induces a subtle change in conformations. The accuracy would be lower when a significant change happens. It is the major limitation of IFD in the current state.
Induced fit is a prevalent phenomenon and it is desirable to include it in molecular docking. There is still a challenge to design a ligand molecule by virtual docking.
But it’s developing. We are always welcome to try new technologies. Please contact us if you are interested in PepMetics® for molecular docking studies.
1) https://doi.org/10.1021/acs.jcim.0c00116
2) https://doi.org/10.1039/c9sc03754c
3) https://doi.org/10.1016/j.ddtec.2013.02.003
4) https://doi.org/10.1371/journal.pone.0058769
5) Free Energy Methods in Drug Discovery: Current State and Future Directions; American Chemical Society, 2021; Chapter 5, pp 127–141
6) https://doi.org/10.1021/acs.jcim.3c00416
7) https://www.charmm-gui.org/