

Advance in Synthetic Data: Current State and Challenges
Data is essential for decision making and its importance is expanding after the emergence of artificial intelligence (AI)-based technologies and methods. Even before the advent of AI, statistical analysis of real data has been widely performed to support the humans’ predictions. In the field of healthcare, however, data collection encounters the challenges of privacy due...
A Rewritten Textbook in Era of Digitalization, Personalization, Collaboration and Innovation
Innovation in technology have brought about paradigm shifts in pharmaceutical science. The landscape of pharmaceutical industry has changed its appearance over the decades, driven by the necessity to adapt itself to consumer expectation, regulatory forces and demographic shifts. It is substantially required for pharma to keep themselves updated and adjusted to the upcoming era so...
TIDES on the Stage: Looking Back on 2023 and Future Perspective
TIDES (peptides and nucleotides) is seeing the light of the day. TIDES gained an intense attention in drug discovery and the number of the approved TIDES by FDA rose up to nine in 2023.1) The molecular entity covers 16% of the approved drugs and it is expanding the realm over the couple of years.2) Still,...
Characterization of Pseudo-Natural Products Synthesized by Fragment Combination
Natural products often have significant biological activity and they have been taken as the pool of drug seeds for long. In order to lead natural products druggable, they were sometimes elaborated and derivatized to increase the PK/PD profile and decrease the toxicity. These synthesized compounds are, so to say, “natural product mimetics”. PepMetics® molecules are,...
Allostery for Target Modulation: Site Prediction and Drug Design
Protein function is modulated not only by competitive inhibition but also by allosteric control. The natural ligand binding site has long been targeted because the mechanism of action is clear and the assay development is relatively easy for the directly targeting drugs. Allosteric modulator of a target protein is serendipitously emerged, but now it is...
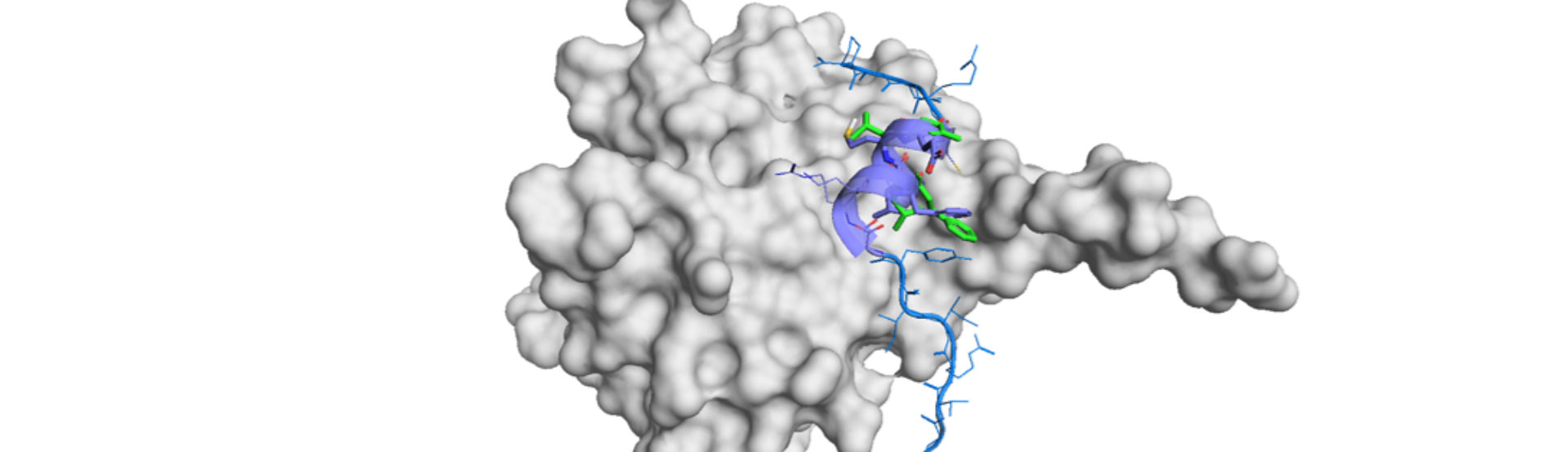
De novo Design and Efficient Synthesis of Orally Bioavailable Small Cyclic Peptides
Cyclic peptides gather interests of many pharmaceutical companies due to the known high potency against various protein targets. The conformationally restricted structure with three-dimensional architecture has enabled us to target proteins called undruggable so far. There are still scarce number of cyclic peptide drug in the market but the number is increasing in this century.1)...
Covalent Inhibitors in Drug Discovery: Current Applications
Aspirin is now known as a covalent modifier of COX-2 through acetylation of Ser530. 1), 2) Since the advent of aspirin in 1899, covalent inhibitors have occupied meaningful range of new drugs. Penicillin, Fluorouracil, Omeprazol, Bortezomib and more covalent drugs have been developed in over 120 years. Even though covalent inhibitors possesses both advantages and...
Integration of Technologies in Drug Design: Molecular Dynamics and Deep Learning
Integration of technologies is a fascinating approach for innovation. A synergy of technologies in high compatibility would increase the success rate in drug design, discovery and development, and reduction of time and cost as well. We are eager to integrate novel technologies in our drug discovery process as the endeavor for the accuracy and acceleration....
Geometric Deep Learning for Structure-Based Drug Discovery
Humans and molecules are living in the same, three-dimensional world. Drug discovery and development is a great work of managing our internal and enemies’ (like virus’) external system by a precisely and optimally elaborated molecule. Molecule-molecule interaction like PPI needs to be analyzed in a three-dimensional world for deep recognition, hence prediction and design of...
Deep Learning Is Enabling Structure-Based Drug Design: Current Approaches
Deep Learning Is Enabling Structure-Based Drug Design: Current Approaches Structure-based drug design (SBDD) or discovery is one of the state-of-art approaches. Computational aid has allowed medicinal chemists to accelerate the drug design in the discovery stages. Now, deep learning (DL) models are enabling the efficient support of SBDD. Emerging DL technologies have potential to lead...