

Published on: January 24, 2024
Protein-protein interactions (PPIs) library is an indispensable and invaluable tool for PPI drug discovery and development. 2P2Idb v21), TIMBAL v22), iPPI-DB3) have been the libraries focused on PPIs and data availability in the recently elucidated cases were limited for long. It will open up to everyone a great opportunity to initiate another PPIs-targeted drug discovery program if well-curated relational database (RDB).
The time has come. DLiP-PPI (Database of Chemical Library for Protein-Protein Interaction) is released in 2023.4), 5) DLiP-PPI is a small-to-medium sized molecule database developed in an AMED project driven by Ikeda group at Keio University and other groups from industry and academia.
The library containing ones over the Lipinski’s rule-of-five. For example, molecular weight ranges from 450 to 650, which is over rule-of-five (MW<500). Library data have been gathered from accumulated in public sources as well as predicted binders of PPI interfaces by 3D docking simulations by FRED software. After filtering a specific range of molecular properties and druggability at a hit stage, and it enabled inclusion of 15,214 molecules at the time of library release.
DLiP-PPI is a web-based, readily available and free library with search and analysis functions. Physicochemical properties, drug-likeness and structure-based search are available. Also, PPI curation search by compound and target is built-in in the web system.
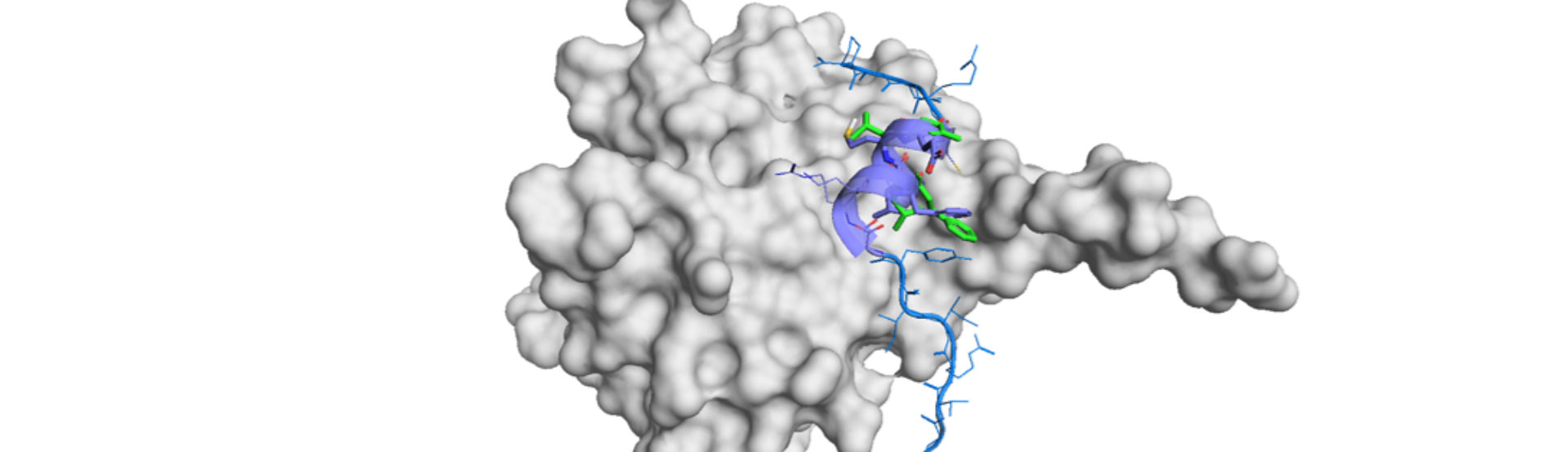
The authors demonstrated the usefulness of DLiP-PPI in combination with machine learning in identification and discovery of inhibitors of Keap1/Nrf2 PPI.6),7) They first performed ligand-based virtual screening (LBVS) and applied two random forest (RT) models, named RF-TI and RF-PI. RF-TI model was generated from truly active and inactive compounds from databases, while PF-PI includes commercial compounds as the putative inactive ones. PF-PI would work as an expansion model in terms of structures and targets. TR-FRET (time-resolved fluorescence resonance energy transfer) assays revealed 15 out of 620 compounds. 5 out of the 15 hits possessed a common substructure and it worked as a hit-expansion methodology.
In the second report, the authors applied DNN (deep learning network) with RF and performed iterative LBVS for improvement. In addition, they switched from RF-TI and RF-PI models to utilization of two different library sets: DLiP1 library for a structure-based drug design, and DLiP2 library for non-flat (sphere-like, spiro, or so) compounds suitable for PPI inhibitors.
First machine learning was conveyed with DLiP1, initial batch of LBVS and TR-FRET assay dataset (FA), the database from the previous work and public data (DB). Then RF/DNN models were performed with these data and DLip2 and TR-FRET activities were measured for selected compounds.
The authors tried five models in this paper and the hit rates ranged 20.0%-27.3%. Compared to random model (1.0%), RF-TI (2.7%) and RF-PI (5.9%), it is obvious that model improvement and iterative approach bumped up the performance of prediction.
The combination of a reliable and target-nature-oriented database and a sophisticated machine learning model produce a broad and expanding opportunity. We will continuously try application of AI-based approaches for drug discovery. We would appreciate it if you are interested in our unique PepMetics library to initiate a collaborative project for PPIs targeted drug discovery and development.
https://doi.org/10.1093/database/baw007
https://doi.org/10.1093/database/bat039
https://doi.org/10.1016/j.drudis.2013.05.003
https://doi.org/10.3389/fchem.2022.1090643
https://skb-insilico.com/dlip
https://doi.org/10.1038/s41598-021-86616-1
https://doi.org/10.1039/D3CC01283B