

Published on: June 5, 2024
Deep Learning Is Enabling Structure-Based Drug Design: Current Approaches
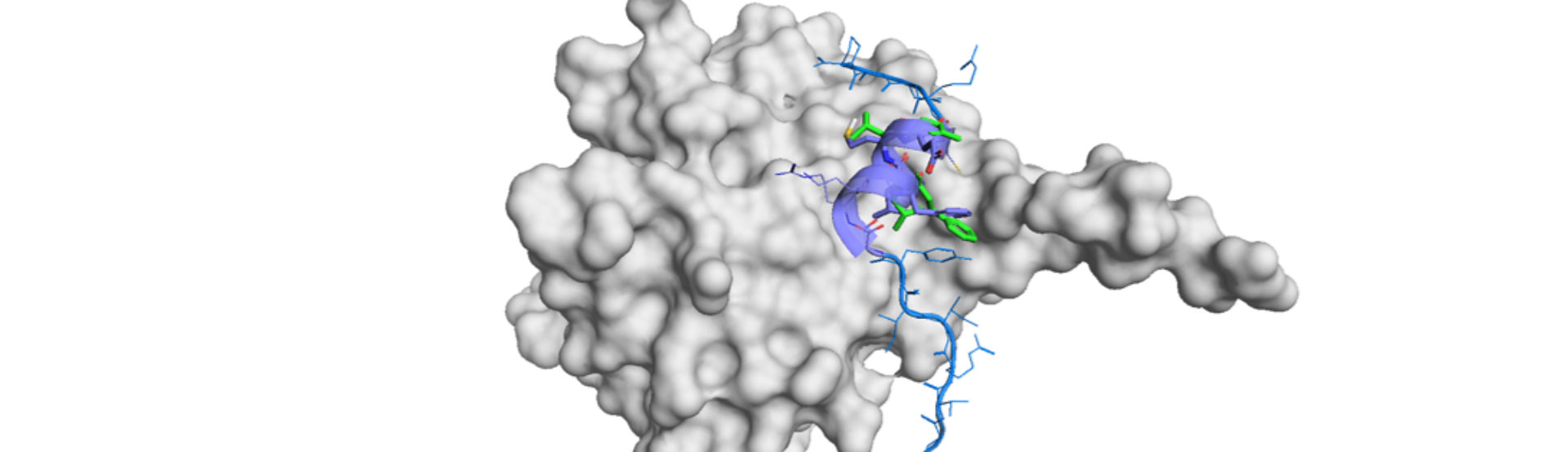
Structure-based drug design (SBDD) or discovery is one of the state-of-art approaches. Computational aid has allowed medicinal chemists to accelerate the drug design in the discovery stages. Now, deep learning (DL) models are enabling the efficient support of SBDD. Emerging DL technologies have potential to lead the stage of drug discovery with the guidance of scientists and engineers.
This review offers us a current stage of DL for SBDD from its basis in the context of drug discovery strategy.1) It includes key terms in the field from the standpoint of both scientists and engineers, and the structural information interpretation in both protein science and deep leaning. Available datasets are also summarized and it would be useful when you start DL approach for SBDD.
In SBDD, recurrent neural networks (RNNs), convolutional neural networks (CNNs) and graph neural networks (GNN) has been mainly employed for every task. The tasks of SBDD consists of at least 3 stages:
-Drug-target interaction (DTI) prediction
-Binding site detection and identification
-Structure-based de novo molecule design
DTI prediction is the starting point of SBDD that requires to predict the biological activity or binding affinity. Sequence-based2),3), 3D structure-based4), 5), and surface-based approaches 6) have been applied by a number of groups. Protein-ligand docking to predict the binding pose of a ligand is also the focused domain of DTI. 7), 8), 9), 10) DL has an advantage on DTI prediction because the molecule complexity does not perturb the accuracy. 11), 12)
A drug discovery projects is often initiated by the library screening against an activity-based assay. Binding site detection and identification is a necessary task to push SBDD forward when a hit compound is found through screening. As in the case with DTI prediction, Sequence-based 13), 14), 3D structure-based 15), 16), and surface-based approaches 17), 18) have been developed so far.
A cycle of SBDD comes to completion when a set of molecules for synthesis and activity test is defined. Structure-based de novo molecule design has been the most challenging tasks, but it is now becoming practical owing to the emergence of generative models based on either ligand or structure.
The possibility of the integrated approaches is expanding owing to the advance in protein structure prediction by DL, AlphaFold2, diffusion model, novel DL approaches like geometric deep learning, and molecular dynamics.
There is still an obstacle of datasets. The scarcity of three-dimensional structural data of homogeneous quality, without missing loop or terminus regions, hampers training of the DL model for structure and DTI and binding-site prediction. Under-reported binding affinities and ADME profiling data is also an issue of the biased training for misleading.
Our AI/DL incorporation strategy is still under developing stage. But we are gathering data in our specific and original chemical space for the intensive application. DL would be a powerful tool for rational SBDD.
Since PepMetics® molecules have different chemical space from conventional ones. We have long been thinking of the specific model optimized for our SBDD approaches.
Please contact us if you have an interest. We would love to show you our specialty and our current state.
1) https://doi.org/10.1002/cbic.202200776
2) https://doi.org/10.1093/bioinformatics/bty593
3) https://doi.org/10.1093/bioinformatics/btz111
4) https://doi.org/10.1021/acs.jcim.7b00650
5) https://doi.org/10.1039/c9sc04606b
6) https://doi.org/10.1016/j.jmgm.2021.107865
7) https://doi.org/10.1093/bioinformatics/btz870
8) https://doi.org/10.7717/peerj.7362
9) https://openreview.net/forum?id=WNwsnE81meC
10) https://doi.org/10.1186/s12859-018-2565-8
11) https://doi.org/10.3390/cells11132117
12) https://doi.org/10.1038/s41598-019-56847-4
13) https://doi.org/10.1021/acs.jcim.1c00475
14) https://doi.org/10.1126/sciadv.aap7885
15) https://doi.org/10.1016/j.sbi.2023.102559
16) https://doi.org/10.1126/science.abj8754
17) https://doi.org/10.1093/nar/gkab1061
18) https://doi.org/10.48550/arXiv.2208.10230