

Published on: 10月 18, 2023
It is no doubt that In silico drug design by structure-based drug design (SBDD) has been a powerful methodology for drug development. Structural data of the target protein, desirably of the binding state with hit or lead compound, delivers us a framework for designing more potent organic molecule structures by medicinal chemists. But with the invent of deep learning, it is realistically worth considering taking advantage of AI-based drug design to figure out a swift and solid approach to reach the candidate.
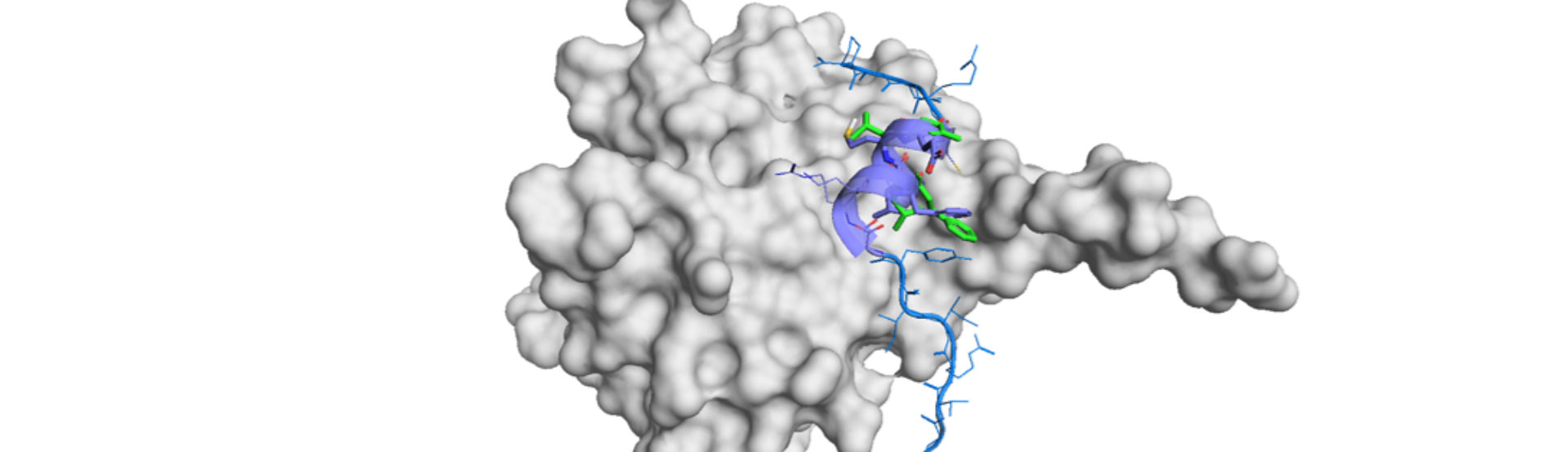
Here is a beautiful demonstration of clear and clever research in communication biology to de novo design of a gastric proton pump (H+,K+-ATPase) inhibitor.1) The authors rationally constructed a series of K+-competitive acid blockers (P-CABs) through the intensive use of deep generative models (DGMs), taking a workflow of Deep Quartet (DQ).2),3) DQ-generated molecules were filtered by computationally and manually and selected ones were synthesized. Then the potent molecule was structurally analyzed by cryo-EM to understand the binding mode against H+,K+-ATPase. SBDD approach improved the affinity and prompt drug development was achieved by the orchestra of three sophisticated technologies.
The group selected the desired pharmacophores by taking the advantage of information of P-CAB binding, represented by SCH28080, soraprazan and tegoprazan. The invaluable and admirable point is that the authors aimed for the generation of a novel and unique molecules that are distinct from the “ancient” P-CABs by the original definition of four pharmacophores,The DQ-generated molecules were evaluated next to figure out the candidates for synthesis. Most of the molecules had moderate synthetic scores (~3)4). The molecules were grouped into three types according to the binding model and just several compounds were synthesized for evaluation. The representative IC50 values were like 39.9±3.3 uM (DQ-02), 33.9±3.8 uM (DQ-04), 49.5±4.9uM (DQ-15), 89.4±13.5uM (DQ-16), 0.70±0.04uM (DQ-06), 292±67uM (DQ-10), 5.57±0.45uM (DQ-11), and 1.54±0.07uM (DQ-12). Although a few compounds showed almost no activity like >2000uM (DQ-09) and >120uM (DQ-14), just one generation by DGMs allowed them to reach two compounds (DQ-06 and DQ-12) that has more potent affinity than the ancient SCH28080 (1.97±0.12uM).
Then Cryo-EM measurement was conducted on DQ-06 and the binding structure was revealed at the resolution of 2.19 Å. DQ calculation had indicated two different binding poses. However, it was unambiguously determined, by the aid of Cryo-EM high resolution structure, to correspond to one of the calculated poses. Thus, they analyzed the binding structure and came up with the hypothesis to introduce a chloride group to enhance van der Waals interaction. Chloro derivatives of DQ-06 were synthesized and revealed that one of the compounds, DQ-18, showed better affinity than the parent molecule (0.31±0.02uM).
This research shows us a powerful framework for de novo drug generation approach for rational drug development. Reasonable molecular design is a difficult step in the early stage. Machine learning has been helped us better selection of molecules to be synthesized with high priority. Now is the age of generation of pretty structures for synthesis.
DGM-based strategy generally succeeds with the related data of high quality. This demonstration was supported by the presence of structural data of the gastric proton pump as well as the P-CABs to figure out the pharmacophores. Structural analysis of PPIs now readily be performed not only by X-ray but also cryo-EM. The collaborative ensemble of DGM, structural biology and synthesis with data accumulation, share and utilization is one of the keys of future drug discovery and development.
The fabulous and rational drug discovery approach was realized by the collaboration of industry and academia, which consists of seven institutes and companies, highlighted by Nagoya University (Abe group: cryo-EM and Yokoshima group: organic synthesis) and Institute for Theoretical Medicine (Dr. Yoshimori: computational chemistry). Collaboration would generate an invaluable opportunity to develop a unique and impressive methodology. Research across the road is necessary for us to bring about an astonishing innovation.
Collaborative research opens up a great opportunity to develop a new strategy and methodology for drug discovery and development. We are eager to make use of PepMetics® and our technologies in any field. We would be glad if we have a chance to collaborate with you to produce an innovative approach.
1) https://doi.org/10.1038/s42003-023-05334-8
2) https://doi.org/10.1248/cpb.c19-00625
3) https://doi.org/10.1002/cmdc.202000786
4) https://doi.org/10.1186/1758-2946-1-8