

Published on: 6月 12, 2024
Deep generative models (DGMs) are changing the story of drug design. They have been applied for drug design in a research level in pharmaceutical companies and still a number of structure-based and ligand-based approaches for generative models are being developed. For recent examples, see 1), 2) for structure-based and 3). 4) for ligand-based ones.
While these approaches are now usable tools in drug discovery, it is critical to optimize the model to each target protein and the hit chemical class to go further into the late stage.
There is always a desire to explore and establish a universal methodology. As one of the DGM-based drug discovery in this context, an interaction-guided drug design framework, including DeepICL (Deep Interaction-aware Conditional Ligand generative model), is reported recently.5)
They only used 10,000 (generally, 100,000~1,000,000) crystal structures for protein curated from PDB to show the generalizability from the side of target-oriented generative drug design.
The authors’ generalization approach has a similarity to that of PepMetics®. We have a similar concept when we work with a partner. Let us see in the article in detail.
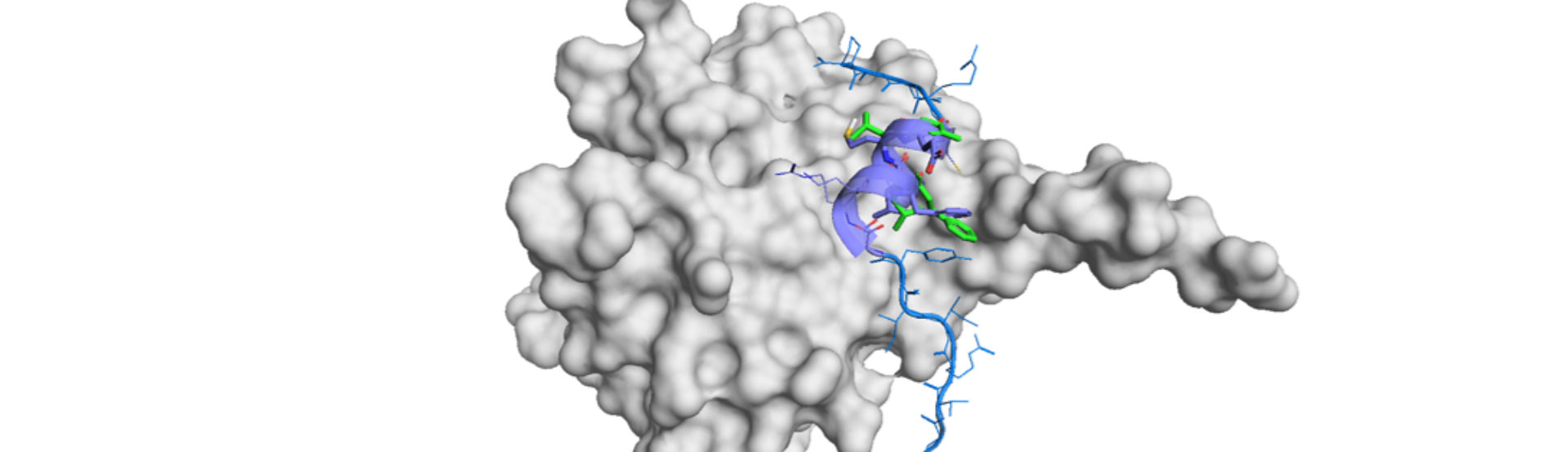
The author developed interaction-aware 3D molecular generative framework, which enables to avoid the dependency on activity data sets. 3D target pocket information is available and predictable now and they utilized it to measure the interaction, or affinity, in an atomic level. Their strategy is separated in two stages.
Stage 1: Interaction-aware condition setting
Stage 2: Interaction-aware 3D molecular generation
The first stage focuses on establishing the interaction condition between the protein binding site and the ligand. Their approach focuses local atom-to-atom interactions instead of the considering entire protein and ligand. They defined interactions by just four dominant interactions and the protein atoms were classified into seven categories for generalization of the residues.
Interactions:
- Hydrogen bonds
- Salt bridges
- Hydrophobic interactions
- πstackings
Protein atoms:
- Anion
- Cation
- Hydrogen bond donor
- Hydrogen bond acceptor
- Aromatic
- Hydrophobic
- Non-interacting
The authors aim to obtain the generality by simplification without loss of the dominating features of interaction and residues in an atomic level. They also use interaction profiler6) when the ground-truth structure of the target is available. The software enables identification of non-covalent interactions automatically from the binding structural data.
The second stage aims for generation of a desirable ligand. Interaction in an atom level drives the molecular generation. In order to predict the reasonable interaction pattens for each protein target, they took reference protein-ligand complexes in a diversified way. The reference data were utilized for the initial core structures, based on visual inspection.
DeepICL is developed to inversely design promising ligands for this dataset. The process is rather simple: DeepICL defines atoms of interest and add atoms one by one to generate a molecule. Thus, DeepICL conserve the critical interactions during generation of ligand structures.
Generalizability of this strategy is demonstrated against mutated EGFR. By defining site-specific conditions between the mutations, Selectivity was represented by the binding affinity scores: -11.8kcal/mol against -3.4 kcal/mol, which correspond to 100-fold difference in inhibitory activity in this case. The author performed another demonstration against Rho-associated protein kinase-1.
DeepICL-based approach is, in the age of AI/DL, one of them. However, you need to understand the necessity of generalization for universal application for any targets. The authors boldly challenged this issue and reached one solution in their logical way.
Our PepMetics® has similarity to Deep ICL because we generally convey collaborative research without disclosing the chemical structure. But the molecule structure is a kind of necessary for us to have a deep discussion. The concept of the stage 1, taking the essence in a scientifically understandable manner, is the way we are dealing with collaborators.
We welcome pharmaceutical companies all over the world for collaboration. We are flexible in terms of the collaboration structure. Please contact us if you are interested in PepMetics®.
1) https://doi.org/10.1021/acs.molpharmaceut.9b00634
2) https://doi.org/10.1021/acs.jmedchem.2c00732
3) https://doi.org/10.1002/jcc.26826
4) https://doi.org/10.1007/978-1-0716-1787-8_12
5) https://doi.org/10.1038/s41467-024-47011-2
6) https://doi.org/10.1093/nar/gkv315