

Drug Design by Sequence Not by Structure of Proteins
Idealistically speaking, it would be fantastic if drug molecule is readily designed just by the sequence of the target of interest. Structure-based drug design (SBDD) is the currently major approach for drug discovery and development stage. The three-dimensional structure is a key resource even though the advent of AlphaFold2 and RoseTTAFold has offered us beautiful...
Can Retrosynthesis Prediction Eliminate Frustration Between Design and Synthesis?
How do you think of computer-based prediction of retrosynthesis? Retrosynthesis is a key in synthesis of any molecule without established synthetic routes. When you design a molecule, it is necessary to try retrosynthetic analysis so as to propose a synthetically accessible one. It is frustrating if the designed molecule...
Possibilities of Macrocyclic Drugs: Expanded by Deep Learning
Macrocyclic drug has been intense research field, represented by macrocyclic peptides.1) It is promising if you see the presence of 67 FDA-approved macrocyclic drugs in 2023. but just over 90 compounds have been reported in approved drugs or those in the clinical stage.2) One of the most examples is the Merck’s phase III case. Merck initiated phase III of MK-0616, an orally available macrocyclic...
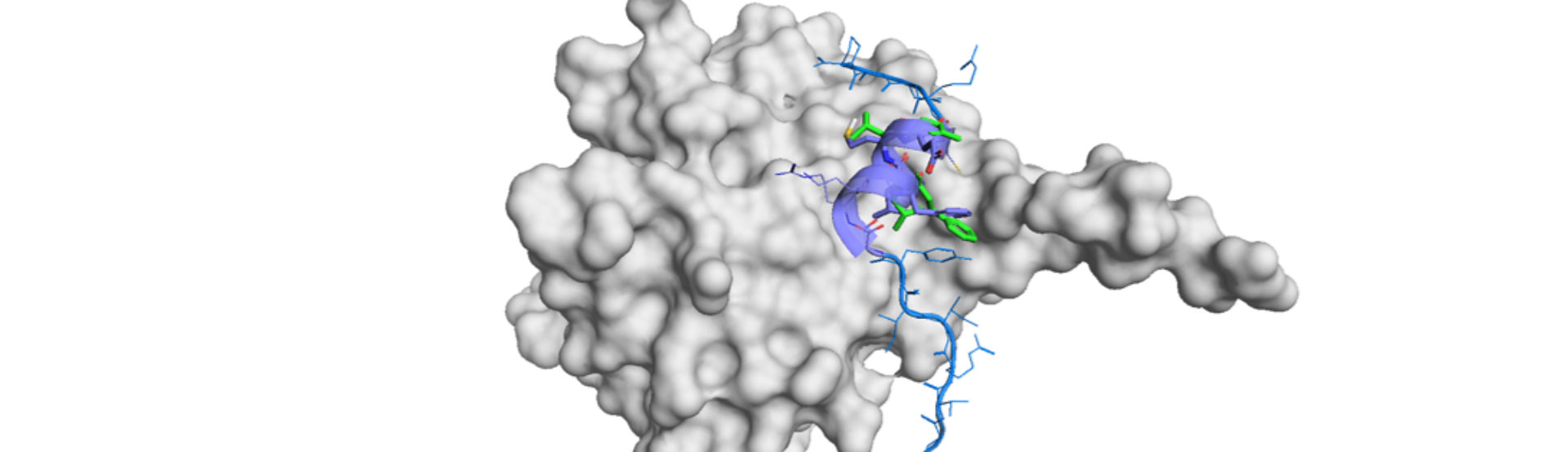
DLiP-PPI library for Keap1/Nrf2 PPI Inhibitor Identification and Discovery
Protein-protein interactions (PPIs) library is an indispensable and invaluable tool for PPI drug discovery and development. 2P2Idb v21), TIMBAL v22), iPPI-DB3) have been the libraries focused on PPIs and data availability in the recently elucidated cases were limited for long. It will open up to everyone a great opportunity to initiate another PPIs-targeted drug discovery program if ...
Recent Alternatives to Improve Permeability of Peptides
Peptide-based drug design is one of the widely investigated field in pharmaceutical sciences for targeting protein-peptide or protein-protein interaction (PPI) inhibitions. Small molecule-based peptidomimetics like PepMetics® is a solution but a biologically active peptide itself is also interesting drug motif for designing a target-specific molecule candidate for a particular disease...
Potential of Stretching Peptides: Mimicry of β-Strands
A small molecule designed for the mimicry of a part of a biomolecule is playing a key role in drug discovery and development. PepMetic® molecules are the mimetics of α-helices and β-turns in a general sense, Mimicry of another well-known secondary structure of proteins, β-strands, would also have a possibility of controlling PPIs. Here is a curious series of papers involving synthesis and characterization of stretching peptides...
Prediction of PPI Affinity: Machine Learning’s Current Situation
Affinity of protein-protein/peptide is significantly important to understand the nature of PPI and to find a target of interest. We are always looking at the PPI binding affinity. Lots of information available on the databases now, owing to tremendous efforts of pharmaceutical companies, academia and research centers in this field. However, affinities are still unknown...
Is Reaction Performance Smartly Predictable by Machine Learning?
Resurgence of machine learning (ML) is becoming a generally usable paradigm shift trigger in a variety of fields. ML has a potential to apply for anything in a sense its artificial neural networks with an appropriate amount of dataset and training mimics human’s way of thinking. In synthetic chemistry, smart and reliable prediction...
Reaction Conditions Optimization: The Current State
In synthetic chemistry, optimization of reaction conditions is a huge task and most of the chemists learn various ways in the university and graduate school as a training. In many cases in the lab synthesis, intuition-based, trial-and-error campaigns are performed when faced with a difficult reaction to maximize the yield, shorten the reaction time, obtain the product with higher purity and so on.
Cyclic Peptide Structure Prediction: Potential of AlphaFold Is Expanding
AI and ITs are offering us fabulous opportunities for peptide structure prediction. The impact of AlphaFold2 and RoseTTAFold on protein structure prediction is the typical example that rapidly changes our attitudes on “prediction”. We are also interested because of the expanding potential of computational approaches. AlphaFold is based on a deep learning method and the structural dataset...