

Published on: 11月 10, 2022
Transition from a macrocyclic peptide to a small molecule with the desired activity is still a huge challenge in drug discovery and development. Here is an interesting strategy “Peptide-to-Small Molecule” developed by Shionogi Pharma Co., Ltd., where small molecules are generated by pharmacophore-guided de novo design from macrocyclic peptides.1
Macrocyclic peptides are goldmines for drug discovery. Numerous efforts have been made to dig out a lead or a candidate from the treasure trove of natural peptides for decades. However, macrocyclic peptides have, in general, low membrane permeability and orally bioavailable drug development have been a serious issue. Simple macrocyclic peptide modification (N-methylation, for instance) would raise the permeability but it is difficult to achieve high affinity and permeability at the same time. From the viewpoint of medicinal chemistry, lead optimization stage has a serious issue due to the limitation of synthetic methodology in the presence of a number of functional groups. It is fascinating if a high-affinity macrocyclic peptide could easily be replaced by a small molecule without significant loss of the affinity and the potency.
Shionogi Pharma’s “Peptide-to-Small Molecule” strategy is a combination of in vitro screening for high-affinity macrocyclic peptide and de novo design of small molecules. They selected peptide display for affinity selection to realize a high-throughput screening (HTS). Shionogi Pharma has nonexclusive technology license agreement with PeptiDream Inc., which has non-natural, synthetic macrocyclic peptide library technology called PDPS (Peptide Discovery Platform System). HTS in vitro screening system obviously matches Shionogi Pharma’s needs to utilize PDPS technology for peptidomimetic drug discovery.
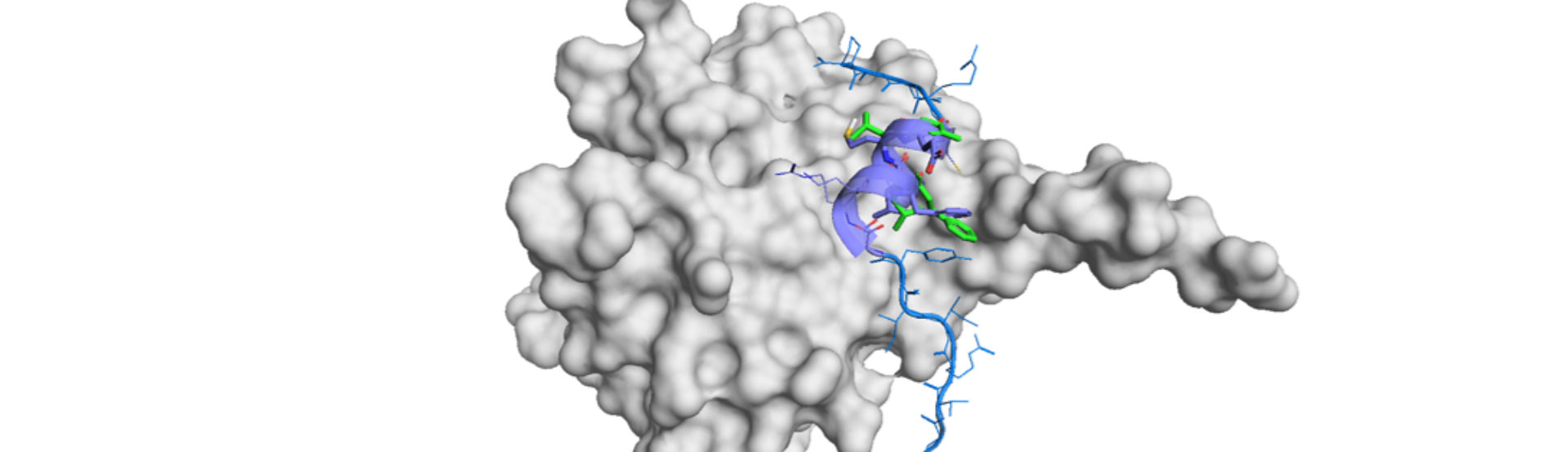
Their de novo small molecule design is guided by pharmacophores, which are figured out by structure-based X-ray co-crystallographic analysis of a target protein and the high-affinity macrocyclic peptide. Potential small molecules would be identified by in silico technologies (i.e., SBDD, virtual screening) and these molecules is readily be designed for synthesis and further optimization. Therefore, their approach is not based on a particular peptide structure but a series of interactions for high-affinity.
The potential of “Peptide-to-Small Molecules” was demonstrated by targeting nicotinamide N-methyltransferase (NNMT). A macrocyclic peptide was identified by cell-free in vitro assay (IC50 = 0.10μM) without any activity on a cell-based assay. Application of their own strategy not only raised cell-free affinity (IC50 = 0.0011μM) but also gave cell-based activity (IC50 = 0.40μM). It is obvious that “Peptide-to-Small Molecules” is a novel option for peptide-based drug discovery.
Thing are changing. There is a review published in 2013, titled “From Peptides to Small Molecules: An Intriguing but Intricated Way to New Drugs” in Current Medicinal Chemistry.2 Strategies from peptides to small molecule was a “Intriguing but Intricated” stage but now it’s becoming reasonable.
Our PepMetics® is a different approach from “Peptide-to-Small Molecules” for PPI drug discovery. PepMetics® is basically structural mimetics: mimicking the backbone and the side chains of a peptide or protein. Shionogi’s strategy is mechanistic mimetics: focusing on the pharmacophore. Their strategy is powerful when X-ray co-crystallographic structure is available while our approach have a potential to realize “Peptide-to-Small Molecule” without a co-crystal. Structural mimetics library has a great potential to find a lead without any information of target-peptide interaction. PepMetics® also provides mechanistic mimetics approach by pharmacophore-guided SBDD and virtual screening. Simultaneous application of both would also be interesting for rapid discovery of PPI drugs, wouldn’t it?
An age of peptidomimetics is coming. Let’s get together to open up new possibility for PPI drug discovery and development by the use of macrocyclic peptides and peptidomimetic technologies.