

Published on: 3月 19, 2024
Idealistically speaking, it would be fantastic if drug molecule is readily designed just by the sequence of the target of interest. Structure-based drug design (SBDD) is the currently major approach for drug discovery and development stage. The three-dimensional structure is a key resource even though the advent of AlphaFold2 and RoseTTAFold has offered us beautiful opportunities. But now drug design by the sequence of protein of interest by the extensive use of deep learning.
TransformerCPI2.0 is a representative and innovative example in this field.1) TransformerCPI2.0 is specialized in connecting protein sequence data with atom vector and atom sequence network.2) The author took advantage of Word2Vec algorithm and self-attention-based encoder TAPE-BERT3) to transform the protein sequence information, and GCNs (graph convolutional networks)4) for atom embedding calculation. The author successfully predicted protein-target interaction in high probability without using protein structure after training their deep learning model by ChEMBL23 database.
The impact of this research is that the authors’ sequence-to-drug approach is demonstrated the performance of deep learning model in case studies in wet experiments. They applied TransformerCPI2.0 in three ways.
1. Evaluation of change in affinity by substitution of a functional group (methyl to trifluoromethyl)
2. Hit finding by virtual screening (speckle-type POZ protein (SPOP) and ring finger protein 130 (RNF130))
3. Drug repositioning by virtual screening of proton pump inhibitor to anticancer drug (ARF1)
The first example showed the influence of -CH3 to -CF3 substitution by their prediction model. Those presented in the paper had consistency with the ground truth at least to see if it results in the activity increase or decrease.
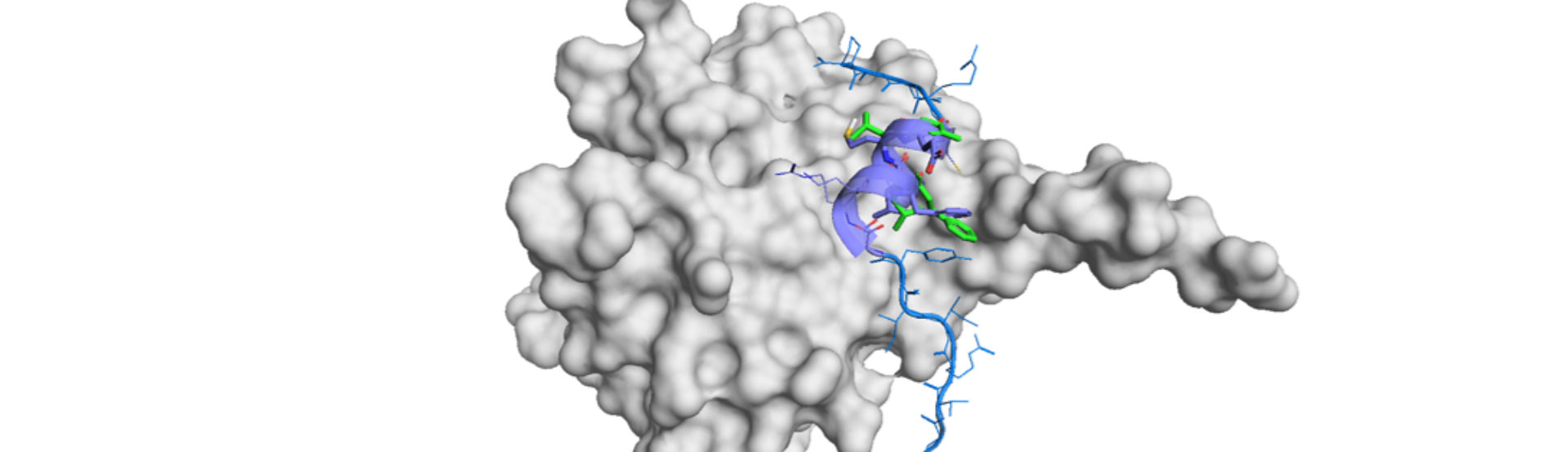
The second demonstration is related to protein-protein interactions (PPIs). SPOP is an adapter protein of cullin3-RING ubiquitin ligase which interacts with the substrate protein.5),6) Just one small-molecule inhibitor is reported7) and the crystal structure of SPOP is not elucidated to date. The author applied their technology to Chemdiv Library8) (1.6 million compounds) and selected top 35,000 compounds by scoring. Then filtering by PAINS (pan assay interference compounds), the automatic clustering of the resultant compounds, and another filtering by Lipinski rules. Finally obtained 82 compounds were subjected to FP assay to find 4 initial hits. But cell permeability was poor for the best hit.
Thus, they conducted hit expansion to 26 compounds, which resulted in 19 hits. They picked the most potent compound with relatively low TPSA so as to overcome the permeability issue. It improved intracellular concentration and the hit, 230D7, were subjected to PK, acute toxicity, and functional assay in cells. The detailed result is summarized in the paper and it had reasonable profile as a hit for drug development.
The third trial is the opposite way of using TransformerCPI2.0. The change of data usage from sequence-to-drug to drug-to-sequence. This inversed approach was applied to drug repositioning of known proton pump inhibitors rabeprazole, lansoprazole, omeprazole and pantoprazole. Sequence-based virtual screening using DrugBank 5.09) datasets identified ARF1 and the following assays determined these compounds act as a covalent inhibitor at C159 of ARF1. The author selected rabeprazole and demonstrated the functional assay in CT26 cells (colon carcinoma cells) and in vivo antitumor effect using colon cancer transplanted tumor models of mice.
Protein targeting by its sequence would open the opportunity of designing and finding a hit against undruggable targets. This technology provides us a possibility for acceleration of drug development story. Wet experiments prove the concept of basic dry research like this sequence-based drug design.
We think it is necessary for all of us to try intensive and collaborative research for innovation. Deep learning-based prediction technologies are advancing in an amazingly rapid pace. We would love to have a chance to collaborate with anyone to develop a practical workflow.
1) https://doi.org/10.1038/s41467-023-39856-w
2) https://doi.org/10.1093%2Fbioinformatics%2Fbtaa524
3) https://arxiv.org/abs/1902.08661
4) https://arxiv.org/abs/1609.02907
5) https://doi.org/10.1016%2Fj.molcel.2009.09.022
6) https://doi.org/10.1038%2Fnature01985
7) https://doi.org/10.1016%2Fj.ccell.2016.08.003
8) https://www.chemdiv.com/
9) https://doi.org/10.1093%2Fnar%2Fgkx1037